Introduction
Over 500 million individuals globally are living with diabetes, with the majority affected by type 2 diabetes mellitus (T2DM).1 The rising prevalence of T2DM is driven largely by modifiable risk factors, including physical inactivity, tobacco use, and excessive alcohol consumption.2 Among the most debilitating complications of diabetes is the development of diabetic foot ulcers (DFUs), which frequently result from a confluence of peripheral neuropathy, peripheral arterial disease, structural foot deformities, and repetitive mechanical stress. These insults are often exacerbated by inappropriate footwear, altered biomechanics, or unrecognized minor trauma.3
DFUs are assessed and classified using a range of clinical criteria, including ulcer size and depth, degree of infection, and vascular and neurological status.4 Despite standardized management strategies, DFUs challenge wound specialists. Between 1% and 4% of individuals with diabetes develop DFUs, and those affected experience a 5-year mortality rate as high as 42%. This mortality statistic is comparable to many malignancies.5,6
The current standard of care (SOC) for DFUs includes serial sharp debridement, biomechanical offloading, reduction of bacterial bioburden, and maintenance of appropriate moisture balance. While these strategies are widely implemented, healing outcomes remain suboptimal. Fewer than 50% of DFUs achieve complete closure within 12 weeks, despite ongoing advances in adjunctive therapies and clinical management strategies.7 The financial burden of DFU treatment is significant, with estimated healthcare expenditures in the United States exceeding $38 million in 2007.8 Socioeconomic status has a well-documented influence on patient outcomes: individuals from low-income or underserved populations often face substantial barriers to accessing advanced wound care services.9 Furthermore, research funding for chronic wounds remains disproportionately low. From 2002 to 2011, only 0.17% of the $7 billion allocated by the National Institutes of Health was directed toward DFU-related research, highlighting a critical gap in national investment relative to the disease burden.10
In recent years, there has been growing interest in the use of cellular, acellular, and matrix-like products (CAMPs) to enhance wound healing. These biomaterials, which include synthetic, biosynthetic, and biologically derived matrices, support tissue regeneration by providing structural scaffolding, modulating the inflammatory response, and promoting cellular proliferation and migration.11 Among these, human-derived allografts, such as amniotic and chorionic membrane products, have shown significant regenerative potential. Amniotic membrane allografts are particularly notable for their anti-inflammatory, antimicrobial, antifibrotic, and pro-epithelialization effects. These properties make them a promising therapeutic option for the treatment of chronic, non-healing wounds.12
Study rationale and design
The present study employs a modified platform trial design to evaluate the clinical effectiveness of two PURION processed lyophilized human amnion/chorion membrane (ppLHACM) products, EPIEFFECT and EPIXPRESS, used in conjunction with SOC, compared to SOC alone, for the treatment of non-healing DFUs. This design is structured under a master protocol, a trial framework that enables the simultaneous and efficient evaluation of multiple interventions within a single, continuously operating clinical trial.13 Platform trials, a subset of master protocol trials, have gained prominence in recent years due to their adaptability and efficiency in comparing multiple treatments across patient subgroups and over time.14
Unlike conventional randomized controlled trials, which are typically limited in scope and time, platform trials incorporate adaptive features that allow for the addition or discontinuation of study arms based on pre-defined interim analyses.15 This flexibility supports real-time decision-making and improves trial efficiency by sharing infrastructure, control arms, and statistical frameworks across multiple investigational products.12 In this study, the adaptive platform structure permits the introduction of additional ppLHACM or advanced wound care products through protocol amendments, ensuring that the trial remains scientifically rigorous and clinically relevant as new evidence and technologies emerge.
By evaluating EPIEFFECT and EPIXPRESS within this robust multicenter, prospective, randomized controlled platform trial, the study aims to generate high-quality comparative effectiveness data to inform the optimal use of biologically derived wound products. Furthermore, the platform approach directly addresses gaps in DFU treatment research by enabling continuous assessment of new therapies in a cost-effective and methodologically sound manner. The findings from this trial are expected to advance evidence-based wound care practices and support the development of dynamic, responsive treatment algorithms for patients with chronic diabetic wounds.
Novel application of platform trial design in CAMP research
To the authors’ knowledge, this study represents the first application of a master protocol, specifically a platform trial design, for the evaluation of CAMPs in the treatment of chronic wounds. While platform trials have been successfully implemented across a range of therapeutic areas such as oncology (e.g., I-SPY 2), infectious diseases (e.g., REMAPCAP), and urgent public health emergencies such as COVID-19 (e.g., RECOVERY and ACTIV-2), their deployment in wound care remains entirely novel and untested in this domain.13,14,16,17
The introduction of this design into the wound care field marks a methodological advancement. Traditional randomized controlled trials in chronic wound management have been constrained by limited sample sizes, single-product comparisons, and prolonged timelines. In contrast, the platform approach enables parallel evaluation of multiple investigational products while leveraging a shared control group to improve trial efficiency and reduce patient exposure to SOC or less effective interventions. Moreover, the adaptive capacity of platform trials allows for the addition or discontinuation of treatment arms based on pre-specified interim analyses, providing a flexible framework that can dynamically incorporate new evidence or technologies as they emerge.
This trial structure addresses a long-standing challenge in wound care research: the need for a scalable, efficient, and scientifically rigorous model capable of keeping pace with the rapid development of advanced therapies. By applying platform methodology to the evaluation of CAMPs, this study establishes a new benchmark for trial design in regenerative medicine. It demonstrates the feasibility of implementing complex adaptive protocols in a field that has historically relied on static, siloed clinical trials, thereby opening new pathways for evidence generation, product comparison, and regulatory advancement in chronic wound therapeutics.
Study objectives
Primary objective
The primary objective of this study is to determine the difference in achieving complete closure of non-healing DFUs with multiple ppLHACMs plus SOC versus SOC alone over 12 weeks using a modified platform trial design. The platform design permits the inclusion of additional products by amending the protocol.
Secondary objectives
There are several secondary objectives of this study, these are:
-
To determine the time to closure over 12 weeks for ppLHACM plus SOC versus SOC alone.
-
To determine the percent area reduction (PAR) at weekly intervals for ppLHACM plus SOC versus SOC alone.
-
To evaluate the frequency and nature of adverse events in subjects receiving ppLHACM plus SOC versus SOC alone.
-
To evaluate quality of life for subjects receiving ppLHACM plus SOC compared to SOC alone using the Forgotten Wound Score (FWS), and standard Wound Quality of Life (wQOL) questionnaires.
Exploratory objectives
The exploratory objectives are:
-
To determine compliance with offloading using digital technology and the effect on wound closure.
-
To determine the proportion of ulcers that heal in patients aged 65 years or older for ppLHACM plus SOC versus SOC alone.
-
To determine the effect of chronic inhibitory bacterial load (CIBL) as assessed by fluorescence imaging (MolecuLight, Toronto, CA) at weeks 4, 8 and 12 on wound closure.
-
To determine the effect of host proteases at weeks 4, 8, and 12 on wound closure (WoundChek, UK).
-
To correlate blood glucose levels (time in range, TIR) to closure using continuous glucose monitoring (CGM) technology.
Material and methods
The study product is sourced from donor materials that undergo a stringent internal screening and testing process. Recovery/acquisition and manufacturing are performed per FDA 21 CFR 1271 and AATB Standards. Processing occurs in an FDA-registered and AATB-accredited facility.
EPIEFFECT and EPIXPRESS
EPIEFFECT is a PURION processed lyophilized allograft membrane derived from human placental tissue and includes the amnion layer, intermediate layer, and chorion layer (Figure 1). It is designed to act as a protective barrier, and is effective for both acute and chronic wounds.18 EPIXPRESS is a PURION processed placental tissue allograft that contains dehydrated human amnion/chorion membrane (Figure 2). The product acts as a barrier and protects the wound bed to aid in the development of granulation tissue in wounds.
FIGURE 1 Image of EPIEFFECT

FIGURE 2 Image of EPIXPRESS

The products are processed using the PURION method, which preserves the human extracellular matrix (ECM) components and is then sterilized. This process allows each layer to be adequately cleansed before processing it into the tri-layer configuration.18 The ppLHACM products are processed by an AATB-accredited facility following FDA current Good Tissue Practice requirements.19
Study design
Overview of study design
This study is a prospective, multicenter, randomized, controlled clinical trial to evaluate two separate ppLHACMs (EPIEFFECT and EPIXPRESS). The study utilizes a modified platform design adopted from clinical trials developed during the COVID-19 pandemic to simultaneously test multiple interventions in a single trial. A trial design diagram is shown in Figure 3.
FIGURE 3 Trial flow diagram. ppLHACM, PURION processed lyophilized human amnion/chorion membrane
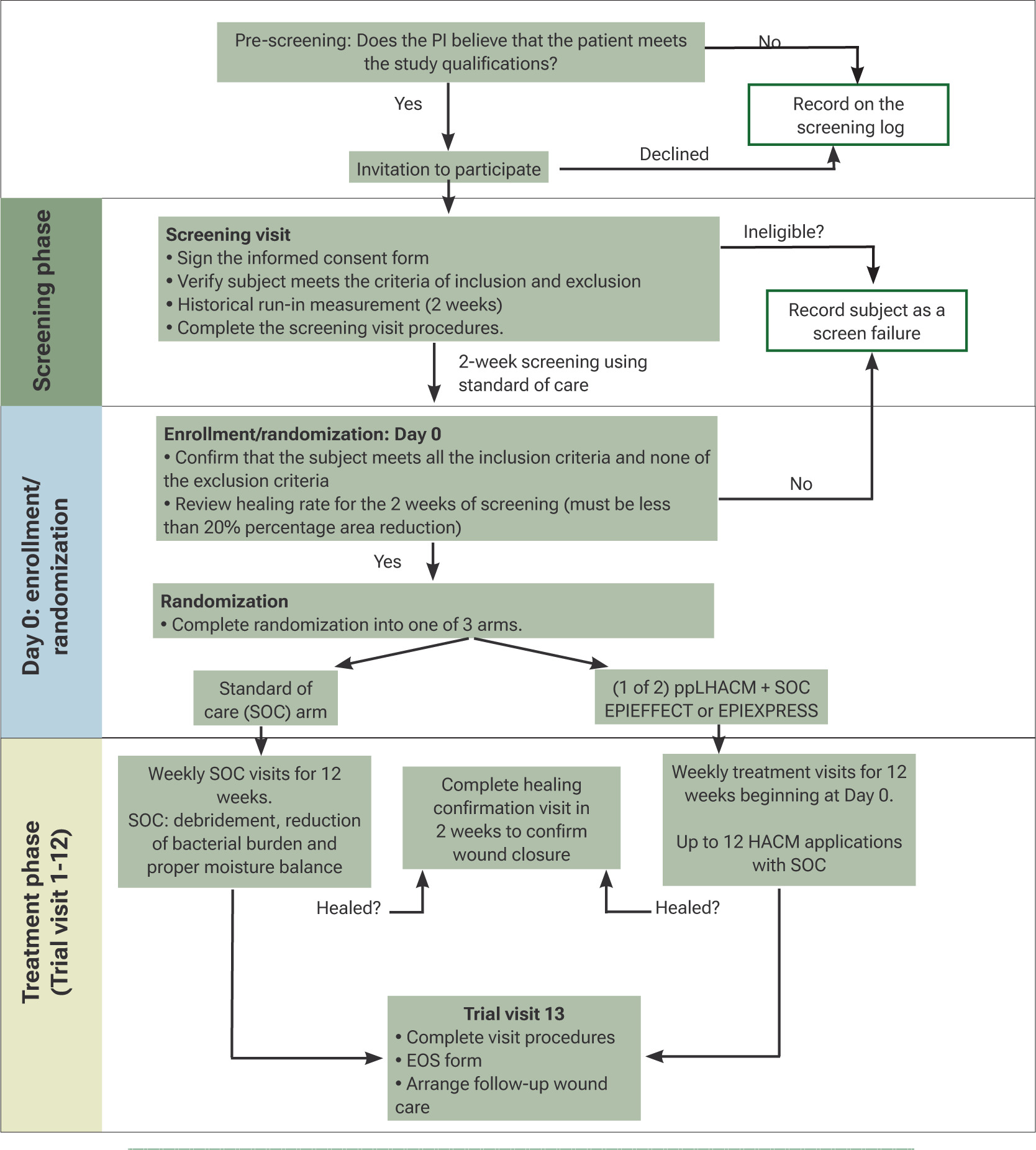
Anticipated duration of the clinical investigation
The study is anticipated to require 24 months to complete. A minimum anticipated recruitment rate of 1.5 subjects per site per month will be completed. To meet the enrollment goals, up to 30 SerenaGroup or affiliated sites will be included.
Evaluation criteria
Clinical endpoints
The primary clinical endpoint will be the percentage of target ulcers achieving complete wound closure in 12 weeks.
Secondary clinical endpoints
The secondary clinical endpoints are as follows:
-
Percentage wound area reduction from trial visit (TV)-1 to TV-13 measured weekly with digital photographic planimetry, using the MolecuLight imaging device, and physical examination.
-
The number of adverse events.
-
Change in pain in the target ulcer assessed using the PEG scale (time frame: TV-1, 3 weeks, 6 weeks, 9 weeks, and 12 weeks or final visit).
-
Change in quality of life using the wQOL (time frame: TV-1, 3 weeks, 6 weeks, 9 weeks, and 12 weeks or final visit).
Exploratory endpoints
The exploratory endpoints are as follows:
-
Compliance with a prescribed offloading total contact casting (TCC) is measured as % of time wearing the TCC as determined by digital technology.
-
Percentage of target ulcers achieving complete wound closure in 12 weeks for subjects 65 years of age or older.
-
Changes in CIBL measured using fluorescence imaging.
-
Changes in host proteases measured using WoundChek EPA test.
-
Correlation between closure and blood glucose levels (time in range, TIR) using CGM.
Study population
The study population will be drawn from patients with diabetes attending wound clinics for treatment of DFUs or minor amputation wound sites. These subjects will be drawn from the general population.
Sample size
The study is intended to provide data to support physician adoption and reimbursement. SerenaGroup performed an extensive literature search on statistical techniques available to power a DFU platform trial.12-14 For a platform design, the allocation ratio is equal to the square root of the number of Intervention groups. This study will include two Intervention groups and one Control group. If additional arms are added (as allowed by the platform trial design), they will enter after the completion of the first interim analysis. Therefore, to maintain 80% power and to detect a difference between groups of 35%, a 1.4:1:1 ratio will be used.
With a total sample size of approximately 170 patients enrolled, the Control arm will have approximately 70 patients, and the two Intervention groups will have 50 patients each. Based on the results of planned interim analyses, the sample size requirements may be re-estimated and adjusted to maintain sufficient study power. The number of subjects may also be increased if additional products beyond the two currently planned are added to the trial as permissible by the modified platform design.
Subject recruitment
Subjects will be recruited from the investigators’ clinical practices within the participating wound care clinics and the general public. IRB-approved social media advertising may be used to recruit potential subjects.
Subject screening (-2 weeks)
Subject screening will be conducted to ensure that subjects meet all the study inclusion criteria and none of the exclusion criteria. Documentation will be required to confirm the ‘historical’ run-in period measurement. A subject is excluded if the surface area of the target ulcer has reduced in size by more than 20% in the 2 weeks prior to the initial screening visit, based on calculating surface area using length X width.
Inclusion/exclusion criteria
Potential subjects will be eligible to participate in the study if the following conditions exist:
Inclusion criteria
-
At least 18 years of age or older.
-
Diagnosis of type 1 or 2 diabetes.
-
At randomization, subjects must have a target ulcer with a minimum surface area of 1.0cm2 and a maximum surface area of 20.0 cm2 measured post-debridement.
-
The target ulcer must have been present for a minimum of 4 weeks and a maximum of 52 weeks of standard of care, before the initial screening visit.
-
The target ulcer must be located on the foot with at least 50% of the ulcer below the malleolus.
-
The target ulcer must be Wagner grade 1 or 2, extending at least through the dermis or subcutaneous tissue, and may involve the muscle provided it is below the medial aspect of the malleolus. The ulcer may not include exposed tendon or bone.
-
The affected limb must have adequate perfusion confirmed by vascular assessment. Any of the following methods performed within 3 months of the first screening visit are acceptable:
-
Ankle-brachial Index (ABI) between 0.7 and ≤1.3;
-
Toe-brachial Index (TBI) ≥0.6;
-
Transcutaneous oxygen measurement (TCOM) ≥40mmHg;
-
Pulse volume resistance (PVR): biphasic.
-
-
If the potential subject has two or more ulcers, they must be separated by at least 2 cm. The largest ulcer satisfying the inclusion and exclusion criteria will be designated as the target ulcer.
-
Target ulcers located on the plantar aspect of the foot must be offloaded for at least 14 days before enrollment.
-
The potential subject must consent to using the prescribed offloading method for the duration of the study.
-
The potential subject must agree to attend the weekly study visits required by the protocol.
-
The potential subject must be willing and able to participate in the informed consent process.
Exclusion criteria
Potential subjects will be excluded from participation in the study if any of the following conditions exist:
-
The potential subject is known to have a life expectancy of <6 months.
-
The potential subject’s target ulcer is not secondary to diabetes.
-
The target ulcer is infected, or there is cellulitis in the surrounding skin.
-
The target ulcer exposes tendon or bone.
-
There is evidence of osteomyelitis complicating the target ulcer.
-
There is an infection in the target ulcer or in a remote location that requires systemic antibiotic therapy.
-
The potential subject is receiving immunosuppressants (including systemic corticosteroids at doses greater than 10mg of prednisone per day or equivalent) or cytotoxic chemotherapy or is taking medications that the PI believes will interfere with wound healing (e.g., biologics).
-
The potential subject is taking hydroxyurea.
-
The potential subject has applied topical steroids to the ulcer surface within 1 month of initial screening.
-
The potential subject has a previous partial amputation on the affected foot that results in a deformity that impedes proper offloading of the target ulcer.
-
The potential subject has glycated hemoglobin (HbA1c) greater than or equal to 12% within 3 months of the initial screening visit.
-
The surface area of the target ulcer has reduced in size by more than 20% in the 2 weeks prior to the initial screening visit (‘historical’ run-in period). MolecuLight imaging device is not required for measurements taken during the historical run-in period (e.g., calculating surface area using length X width is acceptable).
-
The surface area measurement of the target ulcer decreases by 20% or more during the 2-week screening phase: the 2 weeks from the initial screening visit (S1) to the TV-1 visit, during which time the potential subject received SOC.
-
The potential subject has an acute Charcot foot, or an inactive Charcot foot, which impedes proper offloading of the target ulcer.
-
Women who are pregnant or considering becoming pregnant within the next 6 months are excluded.
-
The potential subject has end-stage renal disease requiring dialysis.
-
The potential subject has participated in a clinical trial involving treatment with an investigational product within the previous 30 days.
-
A potential subject who, in the opinion of the investigator, has a medical or psychological condition that may interfere with study assessments.
-
The potential subject was treated with hyperbaric oxygen therapy (HBOT) or a CAMP in the 30 days prior to the initial screening visit.
-
The potential subject has a malnutrition indicator score <17 as measured on the Mini Nutritional Assessment.
-
A subject who has a wound with active or latent infection is excluded.
-
A subject with a disorder that would create unacceptable risk of post-operative complications is excluded.
-
A subject with a known sensitivity to aminoglycoside antibiotics is excluded.
Subject exit/discontinuation criteria
Subjects will exit the study if any of the following conditions exist:
-
The subject voluntarily withdraws from the study.
-
Subject death.
-
Subject acquires any of the listed exclusion criteria.
-
Subject completes the protocol (see Figure 3).
-
Subject is non-compliant with the protocol.
-
Subject’s well-being, in the opinion of the investigator, would be compromised by study continuation.
-
Subject reaches the clinical endpoint of total wound closure (days of wound closure will be counted from the day of closure to day 84).
-
Subject experiences a protocol deviation that, in the investigator’s opinion, will compromise the subject’s continuation in the study.
Study procedures
Informed consent
Subjects will be provided with an informed consent form (ICF) describing the study and providing sufficient information for subjects to make an informed decision about their participation. The consent form will be submitted with the protocol for review and approval by the Institutional Review Board (IRB) and sponsor of the study. The formal consent of a subject must be obtained before he/she is subjected to any study procedure. This consent form must be signed by the subject or a legally acceptable surrogate, and the investigator-designated research professional obtaining the consent. A blank copy of the IRB-approved ICF will be kept on site and by the investigator for review by interested parties.
Vulnerable populations
While vulnerable subjects will not specifically be recruited for this study, vulnerable subjects may be present in the potential subject pool. Additional procedures will not be required to ensure human subject protections for these subjects.
Randomization scheme
Subject randomization will be performed electronically in the GreenLight Guru Clinical electronic data capture system. Subjects will be randomized across up to two Intervention groups and one Control group. The randomization scheme for the Intervention groups is 1:1, with the Control group randomization 1.4:1, depending on the number of Intervention groups enrolling in the study.
Given this is a platform study design, the study may be initiated with two Intervention groups and one common Control group. Following the first planned interim analysis, the sample size may be adjusted to maintain power. Recruitment periods will overlap but will not be identical.
Laboratory testing procedures
Laboratory procedures for this trial include a urine pregnancy test for women of childbearing potential and a fingerstick HbA1c level during screening. A protease swab will be completed on TV1, TV6, TV10, and TV13.
Continuous glucose monitoring (Dexcom)
The Dexcom G7 is a CGM system that provides real-time glucose values to the subject’s smartphone, smart watch, or a Dexcom receiver (provided by the sponsor), and finger sticks are not required. Glucose values will be uploaded into the electronic data capture system at the investigational site. Patients will be instructed on the use of the CGM device by the study site and SerenaGroup, and a coordinator with expertise in managing the Dexcom G7 will be available for consultation.
A large body of evidence has failed to correlate HbA1C values with the healing of DFUs in clinical trials.20 CGM measures glucose values 24 hours, 7 days per week, providing more data points than HbA1c. Recent studies have demonstrated that the amount of time a patient’s glucose values is between 70mg/dl and 170mg/dl correlated with microvascular disease.21 This is called time in range (TIR). TIR has not been studied in DFU clinical trials; however, the investigators are confident that TIR will correlate with the healing of DFUs. The goal TIR is 60%: i.e. the patient’s glucose values are between 70 mg/dl and 170mg/dl 60% of the time.
This is the first DFU clinical trial to utilize this technology; therefore, TIR will not be used to make treatment decisions. A TIR value will also not be used in the exclusion criteria at this time.
Screening visit: target: 14 days before enrollment
During screening, informed consent will be obtained and medical history will be reviewed to determine eligibility based on inclusion/exclusion criteria. If the patient is deemed suitable for the study, demographic information (e.g., height, weight, BMI, gender, ethnicity), medical history, and medication history will be taken.
A vascular screening test will also be taken unless results are available within 3 months of the screening visit. Vital signs will be recorded (blood pressure, pulse, respiratory rate) and a general physical examination will be performed. HbA1c will be obtained and documented (if a HbA1c is available within 3 months of the screening visit the value may be used).
The following assessments will be taken: Mini Nutritional Assessment, Wagner Grade, Fitzpatrick Scale, and Pain Assessment (PEG). With regards to the wound, characteristics will be assessed (amount of granulation tissue, non-viable tissue, depth, exudate, condition of periwound skin). Historical wound measurements from 2 weeks before the initial screening visit will be recorded (‘historical’ run-in period). Digital planimetry (e.g., MolecuLight measurements) is not required for measurements taken during the historical run-in period: calculating surface area using length x width is acceptable. If the wound size has reduced by more than 20% from the historical measurement, the subject will be recorded as a screen failure and will not continue with the screening phase.
Subsequently, the ulcer will be photographed, and the surface area will be measured using the MolecuLight imaging device. Measurements are taken following debridement. Baseline fluorescence imaging will be obtained (MolecuLight procedure, MLiX), and the presence or absence of bacterial fluorescence in the wound bed or on the peri-wound will be recorded.
Begin screening phase (14 days duration leading up to enrollment)
SOC will be implemented and includes the following. The wound will be cleansed using normal sterile saline (NSS). Antiseptics are permitted during the historical screening and 2-week active screening, but not during the treatment phase (except under exemptions listed). Subsequently, sharp debridement will be performed, and calcium alginate or foam dressing provided by SerenaGroup will be applied. Offloading for plantar ulcers or ulcers will be initiated, subject to external pressure. The TCC is the preferred method of offloading. Finally, DEXCOM G7 will be applied, and the patient will be instructed on its use.
TV-1 enrollment/randomization (treatment phase day 0)
14 days after the screening visit (± 3 days), the subject should meet all the inclusion criteria and none of the exclusion criteria. The subject should be assessed for adverse events, medications should be reviewed for any changes, a symptom-directed physical examination performed, and any changes documented.
Additionally, the PAR should be reviewed for the 2 weeks of screening (must be less than 20%), vital signs and wound characteristics recorded, and the Woundchek protease swab procedure should be completed. Subsequently, the ulcer should be photographed and the ulcer surface area measured using the MolecuLight imaging device. Measurements are taken following debridement.
Furthermore, the PEG Pain Assessment, FWS, and wQOL should be carried out. Fluorescence imaging should be carried out and if the image captures bacteria on the wound bed or peri-wound it should be recorded. Glucose levels from CGM should be downloaded, the patient should be instructed on CGM use, and TIR values should be uploaded. If the subject remains a candidate for the trial, continue with randomization; if they do not, it is recorded as a screen failure. Perform SOC and treatment as listed above.
The above procedure is repeated for weekly visits during the treatment phase (TV-2 through TV-12) with the addition of the FWS quality of life questionnaire.
Healing confirmation visit (2 weeks post-wound closure)
Healing is defined by the FDA as complete closure without drainage for 2 weeks. To meet this definition, the patient must return for a healing confirmation visit (HCV) 14 days (±3 days) after the first assessment of 100% reepithelialization. The following procedures will be performed at the HCV: review of adverse events and concomitant medications, assessment of pain status, assessment of closure, wound imaging using MolecuLight imaging device, and confirmation of wound closure.
Withdraw visit
If a subject withdraws early or the investigator terminates subject participation early, the procedures and data collection described for the final study visit (TV-13) will be performed.
Unscheduled visit
Subjects may require unscheduled (UNS) visits (e.g., for dressing changes). The study coordinator will complete the UNS case report form and document the reason for the visit. At an unscheduled visit, the following procedures will be conducted: review of potential adverse events, review of concomitant medications and replacement of secondary dressing, if required.
Follow-up procedures and therapy transitions
At exit from the study, if the wound has not healed, the subject will return to SOC treatments as prescribed by his/her physician. No additional follow-up procedures or transitions are required.
Independent confirmation of healing
In addition to the principal investigator’s determination of complete closure, an independent assessment of the primary endpoint will be performed. Wound care specialists not associated with the trial (‘the reviewers’) will review de-identified digital images of ulcers taken with the MolecuLight device that have healed. Two reviewers who will be blinded to the arm of the study will assess each photograph and determine it as either ‘healed’ or ‘not healed’. If there is a disagreement between the reviewers, the reviewer who agrees with the principal investigator’s assessment will be used.
Data collection and analysis
Subject population(s) for analysis
The following patient populations are subject to study analysis.
-
All-randomized population: any subject randomized into the study, regardless of whether they received study product or treatment.
-
All-treated population: any subject randomized into the study that received at least one exposure to a study product.
-
Protocol-compliant population: any subject who was randomized and received the protocol-required study product exposure.
-
Intent to treat population (ITT): any subject who is randomized according to the randomization assignment.
-
Safety population: any subject randomized into the study that received at least one exposure to a study product.
-
Per protocol population: any subject who completed the study and did not have any major protocol violations.
Statistical methods
The statistical analysis plan (SAP) provides a comprehensive description of the statistical methods and analysis to be used for the evaluation of the data collected in this clinical trial. The hypotheses for each endpoint and planned analysis are provided below.
The primary endpoint is the proportion of wounds achieving complete wound closure in 12 weeks. A chi-squared test of independence is planned. The null hypothesis states that the proportion of wounds achieving complete wound closure in 12 weeks is equal for ppLHACMs plus SOC versus SOC alone. The alternative hypothesis states that the proportion of wounds achieving complete wound closure in 12 weeks is not equal for ppLHACMs plus SOC versus SOC alone.
The secondary endpoint to determine the PAR over weekly intervals for ppLHACM plus SOC versus SOC alone will be assessed using Mann-Whitney testing. The null hypothesis states that the distributions or medians are not equal between treatment groups, while the alternative hypothesis states the distributions or medians are equal between treatment groups.
The number of adverse events and severe adverse events will be reported using descriptive statistics by treatment group. All serious adverse events will be followed and reported.
A linear mixed model will be used to evaluate the quality of life for subjects receiving ppLHACM plus SOC compared to SOC alone using the FWS and standard wQOL questionnaires.
Exploratory endpoints including compliance with off-loading, proportion of target ulcers that heal in patients 65 years or older, chronic inhibitory bacterial load, host proteases, and blood glucose levels will be analyzed using summary statistics and comparison between treatment groups.
Sample size calculation
Based on the assumption that 30% of patients in the SOC group will heal at 12 weeks, a total sample size of approximately 170 patients will be needed to detect a difference of 0.35. The Control arm will have 70 enrolled patients, and the two Interventional arms will have 50 patients each. This will maintain a power of 80% and significance level of 0.05 with a treatment effect of 35%. A two-sided test will be used for all analysis. Sample size determinations were made using the most recent version of R Studio.
Randomization allocation for platform trials
Using Dunnett’s allocation ratio for adaptive design, a study with up to k=2 Interventional groups will have Ök allocation to the standard of care arm. The study will be initiated with two study products and 1 Control group. Additional treatment arms may be added after the completion of the first interim analysis. Therefore, a 1.4:1:1 ratio will used.
Enrolled patients will be randomized using Greenlight Guru and monitored by the study team. Patients will be stratified by wound age and wound size. All strata are listed in Table 1. Patients will be randomized 1.4:1:1 to the Control and Intervention arms, respectively.
TABLE 1 Strata for randomization.
| Variable | Strata | Groups |
|---|---|---|
| Wound age | <60 days | 1. <2cm2 wounds <60 days old 2. <2cm2 wounds >60 days old 3. 2-3cm2 wounds <60 days old 4. 2-3cm2 wounds >60 days old 5. >3cm2 wounds <60 days old 6. >3cm2 wounds >60 days old |
| >60 days | ||
| Wound size | <2cm2 | |
| 2–3cm2 | ||
| 3cm2 |
Interim and final analysis
An interim analysis is planned when a minimum of 25 patients in each Interventional arm have completed care (study products 1 and 2), and a minimum of 35 have completed SOC.
Intervention arms will be dropped if mean PAR per intervention arm is less than 10% at the interim analysis. Should a treatment arm meet the removal threshold, an investigation will be conducted to determine if factors other than product performance impacted the clinical results.
The study will continue enrolling patients until the Control arm reaches approximately 70 patients and the 2 Interventional arms reach 50 patients each. Additional interim analyses may be conducted based on pre-determined enrollment targets as defined in the statistical analysis plan.
Definition of percent area reduction
PAR is the percent change in the wound surface area of the index ulcer and will be calculated using the following formula:
Where A1 is the baseline area (at randomization), and A2 is the area at the specified time point.
Poolability of treatment arms
During interim analysis, tests to determine if the treatment arms can be pooled will be conducted. If these tests are not significant, treatment arms will be analysed individually against the SOC.
Anticipated benefits and risks/risk mitigation
The benefits associated with this study are that investigators will gain insight into the safety and performance of the ppLHACMs being evaluated. Risks associated with ulcer care related to this study include ulcer management and wound cover risks. Potential risks associated with study procedures are listed in Table 2 along with the applicable risk mitigation.
TABLE 2 Potential risks and risk mitigation.
| Study procedure | Anticipated risks | Risk mitigation |
|---|---|---|
| Wound debridement | Pain | Procedures to be performed by trained clinical staff |
| Wound measurements | None anticipated | N/A |
| Pain assessments | None anticipated | N/A |
| Wound photos | None anticipated | N/A |
| ppLHACM/skin substitute application | This allograft has the potential to transmit infectious disease to the recipient Potential allergic reaction/skin irritation |
Strict donor screening and laboratory testing, along with dedicated processing and sterilization methods, are employed to reduce the risk of any disease transmission. However, as with all biological implants, an absolute guarantee of tissue safety is not possible Patients with a known sensitivity to aminoglycoside antibiotics are excluded from participating in the study |
| Dressing placement | None anticipated | N/A |
| Total contact casting | None anticipated | N/A |
| Ankle-brachial Index (ABI) | Discomfort in area of skin breakdown secondary to pressure from cuff | Topical lidocaine |
| Continuous glucose monitoring (CGM) | Local skin irritation, failure of the CGM to function properly, and inaccurate glucose measurements | Patient education and weekly monitoring. Obtain serum glucose and or fingerstick glucose if inaccuracy of the CGM is suspected |
To minimize the risks during the study, the investigators have been chosen for their qualifications by training and experience. All procedures will be performed on subjects who are candidates for chronic ulcer management. Subjects will be carefully monitored throughout the follow-up period. Adverse events and adverse reactions will be documented and evaluated during follow-up visits.
Discussion
DFUs represent a significant global health burden, affecting a substantial proportion of individuals with type 2 diabetes and contributing to increased morbidity, mortality, and healthcare expenditures. Despite the availability of established SOC practices, including sharp debridement, pressure offloading, infection control, and moisture management, clinical outcomes remain suboptimal. Fewer than half of DFUs achieve closure within 12 weeks of treatment initiation.7 These healing challenges are compounded by socioeconomic disparities in access to advanced wound care, underfunding of chronic wound research, and a scarcity of high-quality comparative effectiveness data to guide therapeutic decision-making.8–10
In response to these unmet needs, interest in the use of CAMPs has increased. These advanced wound care products have demonstrated favorable biological activity in preclinical and early clinical studies and may enhance healing in complex, non-healing wounds. However, their evaluation has been limited by the constraints of conventional randomized controlled trials, which often assess single products in isolation, without accounting for evolving clinical standards or permitting comparative assessment of multiple interventions within a consistent framework.
This study introduces a novel approach to CAMP evaluation using a modified platform trial design under a master protocol. This is the first known application of such a design in chronic wound care research. Platform trials have proven highly effective in therapeutic areas such as oncology and infectious diseases, where they have accelerated therapeutic discovery, improved trial efficiency, and enabled adaptive refinement of interventions based on emerging data.13–15 Applying this approach in wound care marks a significant methodological advancement by providing a flexible and scalable infrastructure for evaluating multiple biologic products in parallel against a shared SOC comparator.
This design offers several advantages. It improves resource utilization using shared trial infrastructure, centralized data management, and a common control arm. It enhances statistical power and generalizability by supporting the simultaneous evaluation of diverse product candidates. Furthermore, its adaptive features allow for protocol modifications based on pre-specified interim analyses, enabling the addition of new investigational products without requiring separate trial initiation. These attributes collectively address long-standing limitations in wound care research, particularly the inefficiency and fragmentation of traditional clinical trial designs.
Beyond its methodological innovation, the study will generate important clinical data on the effectiveness of two ppLHACM products, EPIEFFECT and EPIXPRESS, used as adjuncts to SOC in the treatment of non-healing DFUs. The multicenter, prospective randomized trial design will support robust assessment of clinical outcomes and contribute valuable evidence to inform regulatory policy, clinical practice guidelines, and payer decision-making in advanced wound care.
Another unique feature of this trial is the integration of CGM technology, which represents an innovative advancement in the design of DFU clinical research. Glycemic control is a well-established determinant of wound healing outcomes in individuals with diabetes, yet traditional measures such as point-of-care fingerstick glucose and HbA1c provide only intermittent or retrospective assessments of glucose variability. CGM offers real-time, dynamic tracking of interstitial glucose levels, allowing for a more granular understanding of glycemic excursions, TIR, and hypoglycemic events. By incorporating CGM into this trial, investigators can assess the association between glycemic fluctuations and wound healing trajectories with greater precision than previously possible. Moreover, CGM data may help identify patients at risk of delayed healing due to glycemic instability and provide insight into how interventions targeting glucose control can be optimized in parallel with advanced wound therapies. To the authors’ knowledge, this is the first prospective DFU trial to systematically incorporate CGM technology as both a clinical monitoring tool and an exploratory biomarker for treatment response, establishing a precedent for the integration of metabolic data into wound care research.
Conclusion
This trial is expected to provide high-quality efficacy data on ppLHACM products and contribute to evidence-based practice in diabetic foot ulcer management. The novel modified platform design offers a flexible and efficient approach to evaluating multiple interventions within a single clinical trial.
Future research may expand on this platform to include additional product classes, explore subgroup analyses based on wound characteristics or patient risk profiles, and incorporate emerging digital technologies for wound monitoring. The successful implementation of this trial may serve as a model for future investigations in regenerative medicine and support the evolution of more responsive, efficient, and evidence-based approaches to chronic wound management.